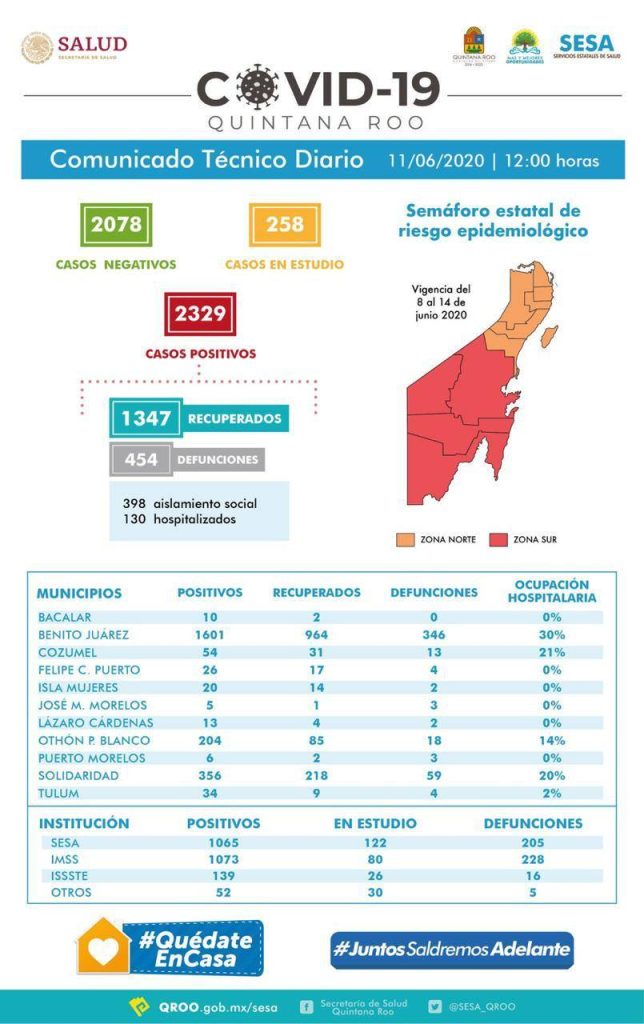

El virus que causa el COVID-19 podría tener un punto débil en su interior: una enzima indispensable para su supervivencia y sobre la que ahora se conocen nuevos detalles, lo que puede servir para el desarrollo de fármacos, según un estudio que publica este martes la revista Chaos.
El científico español Ernesto Estrada, que trabaja en el Instituto Universitario de Matemáticas y Aplicaciones de la Universidad de Zaragoza, firma este estudio centrado en la proteasa (enzima que rompe los enlaces de las proteínas) principal del coronavirus.
Esta proteasa principal sería “como el aparato digestivo, la que realiza la transformación de las poliproteínas en el virus”, explica Estrada.
Estrada ha descubierto un aumento importante en la sensibilidad de la proteasa principal del SARS-CoV-2 frente a pequeñas alteraciones que puedan cambiar su estructura, en comparación con la del coronavirus SARS-CoV-1, que apareció entre 2002 y 2003.
El científico estudió cómo podría usarse ese aumento de la sensibilidad para que inhibidores de la proteasa (moléculas orgánicas, medicamentos o nuevos compuestos químicos) pudieran atacarla, ya que sin ella el virus no puede sobrevivir.
Los resultados del estudio abren la puerta para usar protocolos de detección masiva de moléculas que permitan identificar potentes inhibidores de la proteasa principal del SARS-CoV-2 y, por consiguiente, para el desarrollo de nuevos medicamentos.
La proteasa principal en los coronavirus no suele cambiar tanto como puede hacerlo la proteína Spike (S), que es la que usan estos virus para introducirse en las células humanas. “Esa es una de las ventajas de atacarla”, dice Estrada.
La otra es que no existe una proteasa semejante en los humanos, por lo que la especificidad de los fármacos puede ser muy significativa.
Estrada analizó de cerca la proteasa principal del SARS-CoV-2, lo que le permitió descubrir diferencias con la del SARS-Cov-1, a pesar de que ambas tienen un 96 % de semejanza en sus secuencias de aminoácidos.
Usando técnicas matemáticas estándares, estas diferencias sutiles son imperceptibles por lo que tuvo que usar técnicas más avanzadas desarrolladas por él y su equipo hace un par de años.
El investigador español tiene experiencia en el análisis matemático de redes, por eso decidió tratar esa enzima como si de una red se tratara y, para ello, representó su estructura de una manera matemática, en la que cada uno de sus 303 aminoácidos es un nodo y la interacción física entre ellos es representada con aristas.
Se dibuja así una especie de “telaraña” que representa a la proteasa principal del coronavirus.
Cuando en una telaraña cae un insecto, se genera una perturbación en el tejido que avisa al arácnido del lugar en el que se encuentra la presa. Una telaraña sería más efectiva cuanto más sensible sea transmitir esas perturbaciones y cuanto más lejos se transmitan.
Si se compara “la telaraña” del coronavirus de 2003 con la del actual, se ve que esta última es casi un 2.000 % más susceptible a la transmisión de esas perturbaciones, que pueden producirse por cualquier interacción molecular en el ambiente intracelular.
Además, la proteasa del SARS-CoV-2 es capaz de transmitir esas perturbaciones mucho más lejos que lo que lo hace la del CoV-1 a través de la proteína.
Siguiendo con el símil de la telaraña, Estrada indica que las presas que caen en ella serían el análogo de los inhibidores, los cuales se depositan siempre en un “lugar” muy concreto, un pequeño grupo de aminoácidos, que rodean al sitio activo de la proteasa con el que la enzima realiza su función.
Estrada estudió tres tipos de inhibidores, dos de ellos “extremadamente potentes”, y descubrió que en la medida que un inhibidor “sea capaz de reducir lo menos posible esa sensibilidad a la transmisión de perturbaciones, más efectivo va a ser contra la proteasa”.
Esa mayor sensibilidad a las perturbaciones hace que la proteasa sea más eficaz en su trabajo como enzima, pero, a la vez, deja una puerta abierta que “puede ser un talón de Aquiles” a favor de los inhibidores.
Fuente: EFE